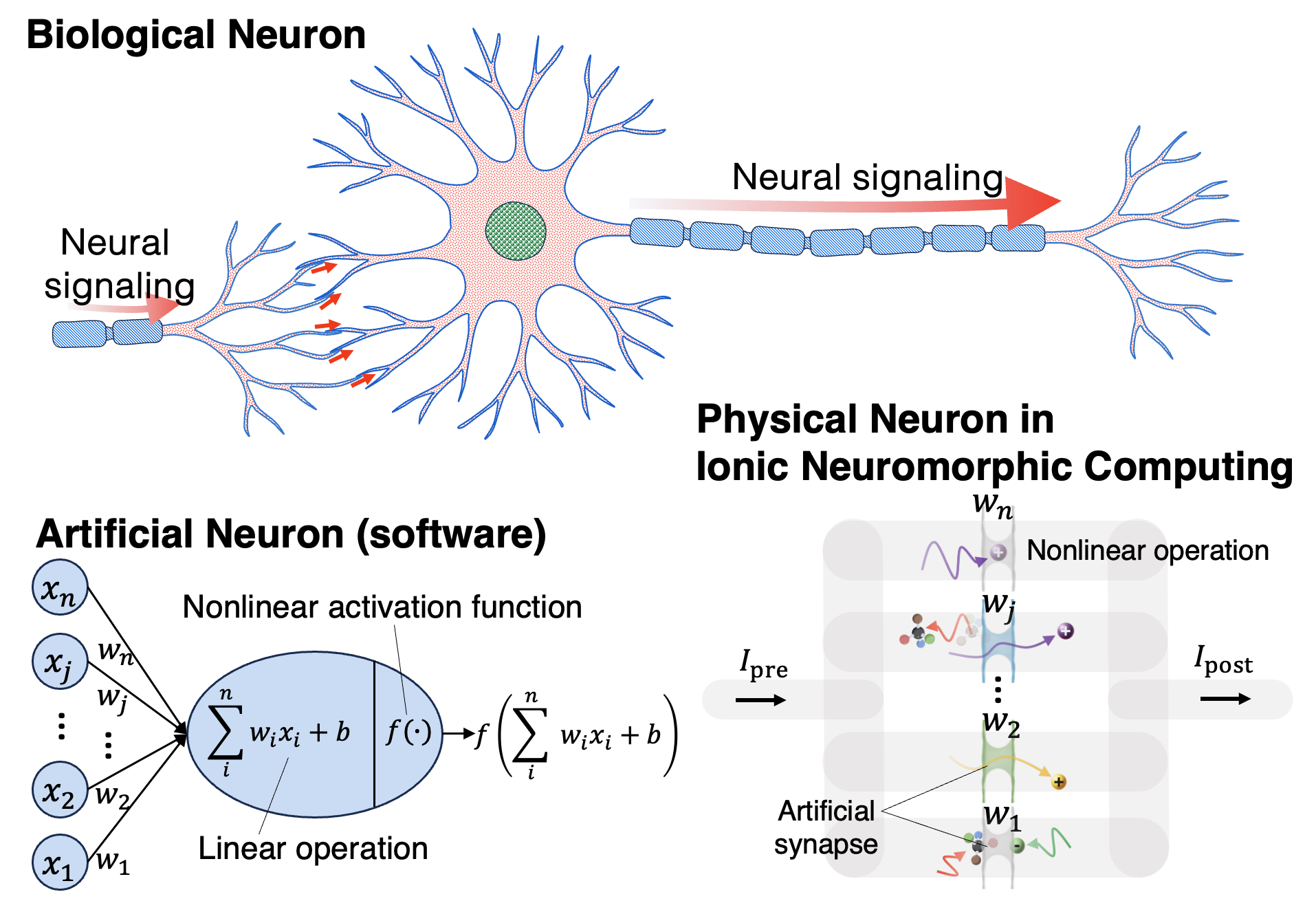
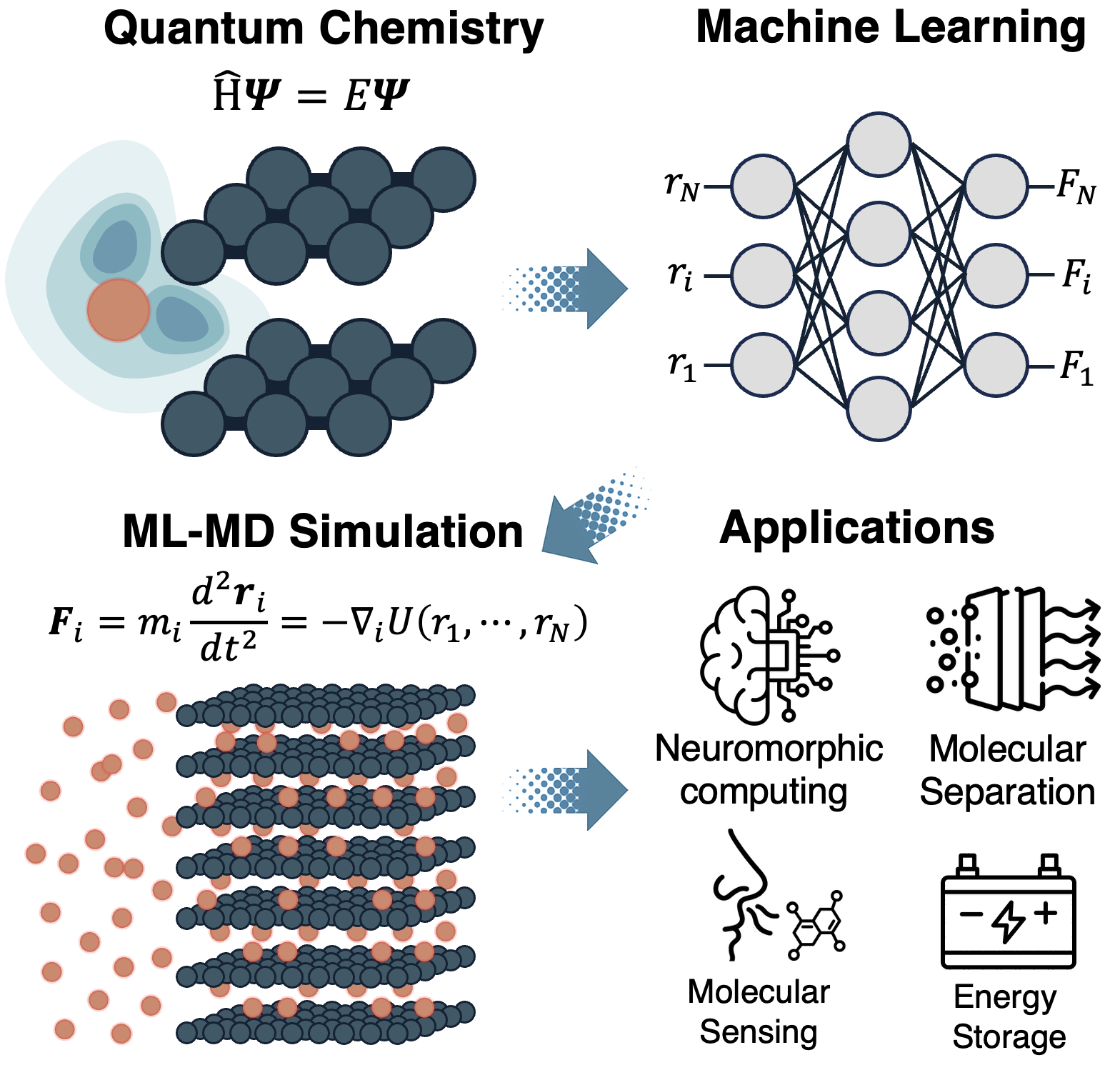
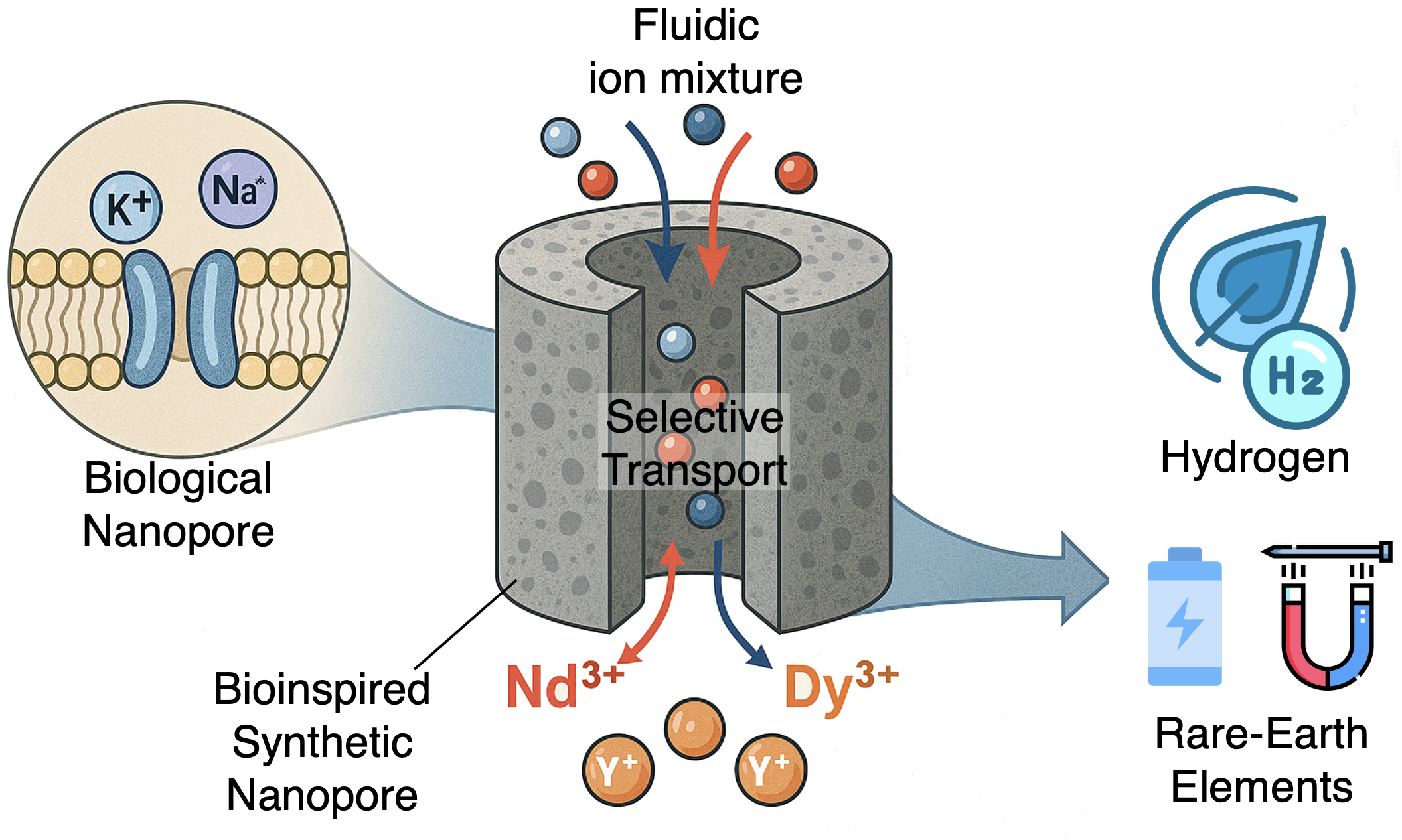
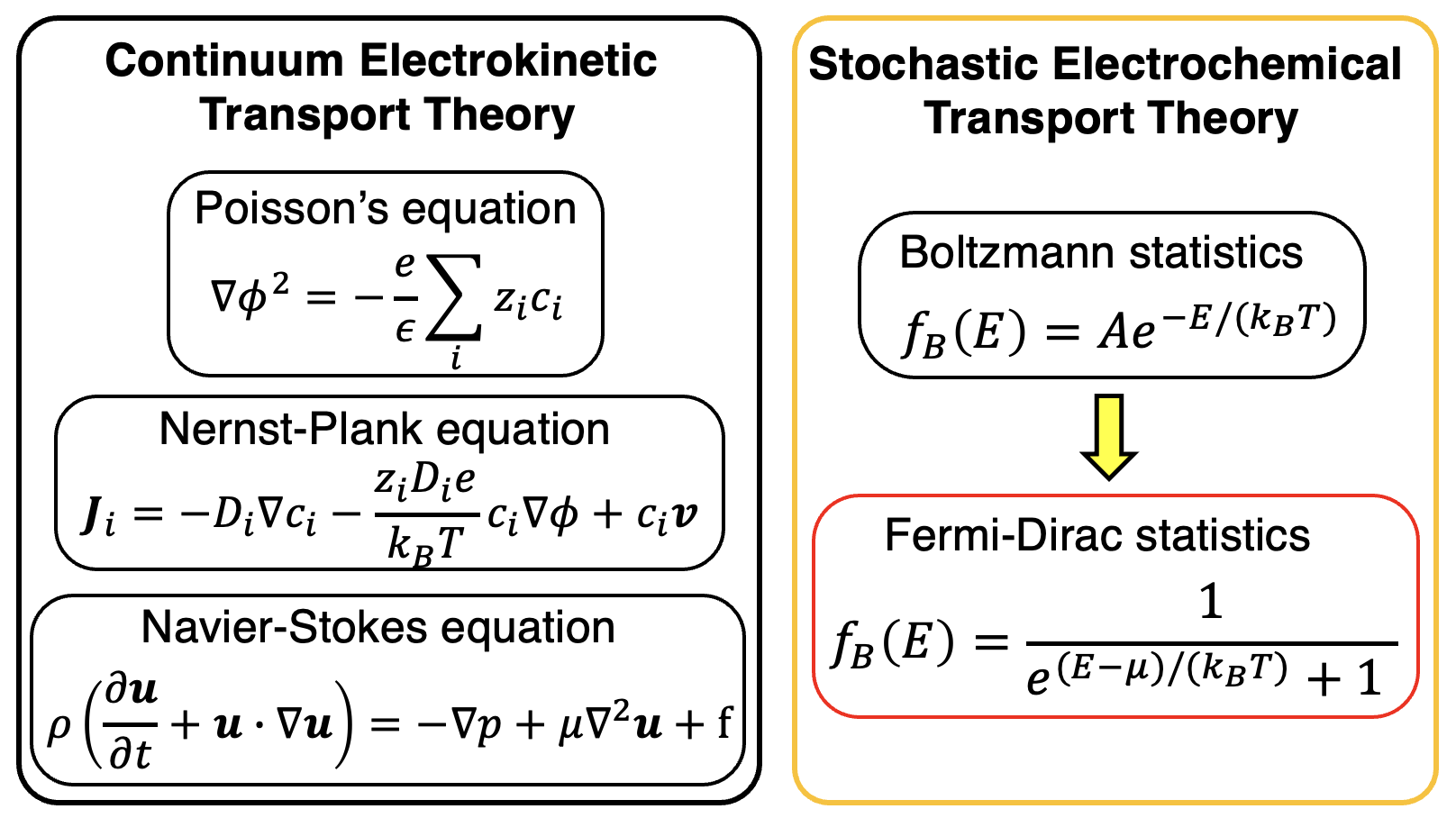
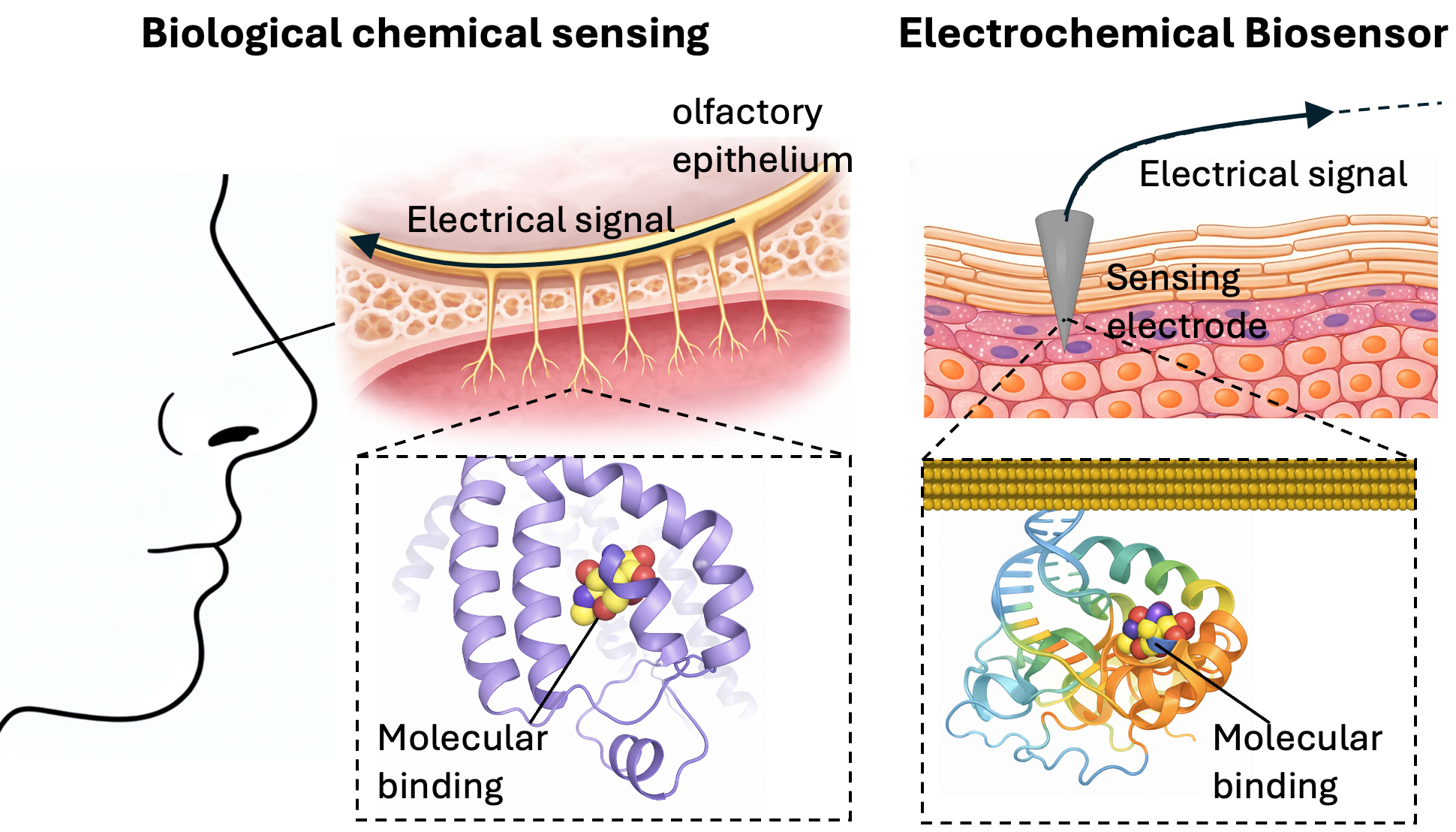
Biomimetic Nanofluidic Chemical Sensors
We develop computational screening tools for electrochemical affinity (EA) biosensors to accelerate the detection of diverse disease biomarkers at the point of care. By integrating GPU-accelerated molecular dynamics simulations with transformer-based deep learning, we build interface-aware ML prediction models for interfacial conformation and binding affinity, enabling automated, high-throughput screening of large aptamer libraries against diverse biomarkers under realistic electrode-interface conditions.
Sub‑projects
- Interfacial Aptamer Conformation: All-atom MD simulations of DNA aptamers tethered to Au electrodes to understand how electrode interfaces influence conformation and binding.
- Analyte Accessibility: Free energy calculations to quantify how electrode interfaces influence target availability near tethered receptors.
- Interface-Aware ML Framework: Transformer-based deep learning to predict electrode-tethered aptamer conformations without new MD simulations for each candidate.